Mar 19, 2020 | Lung Disease
by Elise Wachspress
Ask just about any adult if they carry a scar, and most will admit they do. Some will point to the skinned knee that healed over badly or a rakish cleft like that in Harrison Ford’s chin; perhaps the line from a long-ago surgery or the vestige of childhood chicken pox left on an otherwise flawless décolletage. As Cormac McCarthy said, “Scars have the strange power to remind us that our past is real.”
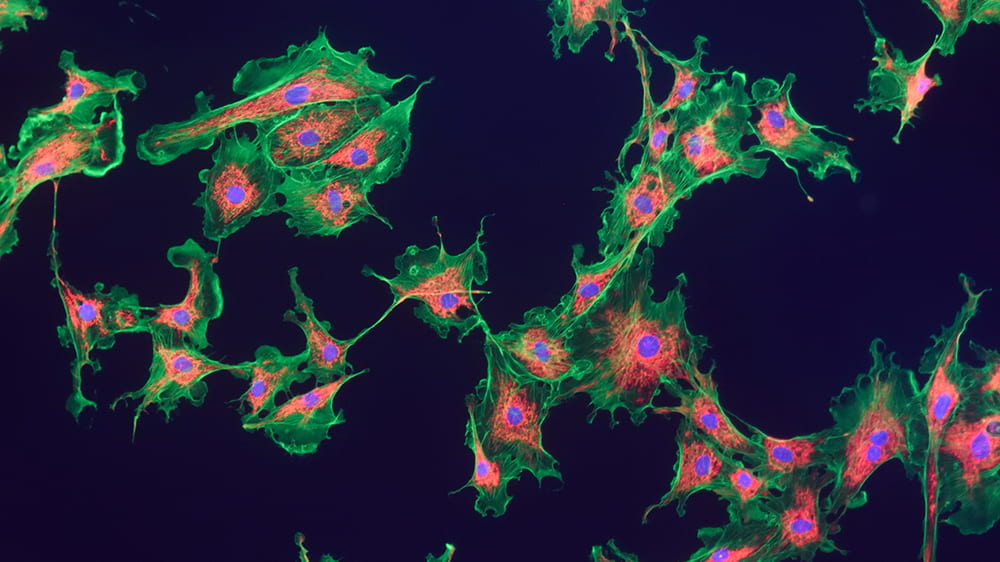
Scars are actually webs of collagen. An injury leads to a healing response from multiple types of cells, including myofibroblasts, which manufacture the collagen that holds cells back together. Though scars may recall a painful moment in our past, they also restored our protective envelope of skin, leaving us healthy, though a little dinged.
But what if scarring becomes progressive, most often in our internal organs? In fibrotic disease, continued insults to the system lead to a runaway response, and the myofibroblasts don’t stop making collagen. The tissues involved get increasingly rigid, excess collagen starts encroaching on healthy cells, and eventually the organs involved begin to fail.
Progressive fibrosis is usually a slow process, making it hard to tell exactly what trigger or combination of triggers set the whole process in motion. This is often a particular mystery in the case of pulmonary (lung) fibrosis. With every breath, we take in everything from mold, bacteria, and viruses to environmental chemicals: which of these is the culprit? And as with allergies to substances like pet dander or peanuts, not every person responds aggressively to these factors, thus individual genetics are likely to be a factor in the disease as well.
Unfortunately, pulmonary fibrosis is fatal, and current therapies are not effective. Researchers are thus highly motivated to tackle this confoundingly complex problem from many angles. While many are particularly interested in the cell signaling that guides the process, Gokhan Mutlu, MD, and Robert Hamanaka, PhD, have been looking to starve the beast: they want to understand what nourishment myofibroblasts need to make the collagen.
Like a complex chemical plant, our bodies take in a variety of raw materials, refashion these into new chemicals, and finally manufacture the many specific compounds (proteins, lipids, nucleic acids) each of our cells need to survive and thrive. And like most chemical plants, our bodies depend on enzymes to lower the activation energy needed to get each of those chemical processes started. Mutlu and Hamanaka want to parse out the many steps in the process—from raw materials to pathological cellular outcomes—to find more than a few places to intervene in the production line and short-circuit the disease mechanism.
Knowing an amino acid called glycine makes up about a third of collagen’s structure, Mutlu, Hamanaka, and their team tried reducing this amino acid in the diets of their mouse models. But this didn’t work—it appears the fibroblasts prefer to make the glycine themselves.
They next turned to intervening in what is known as the “serine-glycine synthesis pathway,” to see if that might slow the whole fibrotic mechanism. The pathway is an important step in converting glucose—the most common carbohydrate circulating in our blood and energy source for our bodies—into the building blocks used for collagen production.
Mutlu and Hamanaka identified an enzyme crucial in the process: by using a drug to inhibit the enzyme, they could slow conversion of glucose into glycine in mice—a finding important not only in pulmonary fibrosis, but potentially in other types of organ fibrosis, like cirrhosis of the liver, as well as cancer.
Continuing to explore other parts of the collagen production process, they looked to see if they might intervene in the conversion of glutamine into proline, another amino acid essential to the collagen-making process. Proline, it turned out, also needed to be made fresh within the cells—ingesting glutamine and proline in food or as a drug did not seem to spark the collagen production. And just as with the first enzyme they identified, this second also may have relevance for many types of organ fibrosis and cancer.
This work again demonstrates how basic science—understanding how the body works and the many ways our metabolic processes can go wrong—can inform new treatments in multiple diseases.
And perhaps makes many of us grateful that our scars are evidence of the past, and not ongoing events.
Elise Wachspress is a senior communications strategist for the University of Chicago Medicine & Biological Sciences Development office

Nov 8, 2018 | Lung Disease, Research
by Elise Wachspress
Stop for a minute and breathe, just breathe. Now envision what your lungs are doing.
In your mind’s eye, do you see big pink balloons filling and deflating?
Close. What’s really going on is hundreds of millions of tiny little pink balloons filling and deflating—ten to twenty times a minute, even more often for babies and small children. Your lungs actually look more like foamy, pulsating soap bubbles than balloons.
In between these bubbles—the alveoli—is a flexible web that allows the little sacs to expand and contract, taking in air, grabbing the oxygen, and dispelling the carbon dioxide waste products.
Unless the web starts to harden. That’s what happens in the case of interstitial lung diseases (ILD). Alveolar expansion gets more and more difficult, until patients begin to feel their lungs are made of concrete. The damage is usually progressive and irreversible. In some cases, certain medications can slow ILD, and some people may be candidates for lung transplants. Otherwise, it’s not hard to see where this condition leads. When you can’t breathe, you can’t live.
ILD can be caused by exposure to hazardous materials like asbestos. Sometimes autoimmune diseases, like rheumatoid arthritis, can trigger the disease. Unfortunately, in most cases, especially for a subset of the disease called idiopathic pulmonary fibrosis (IPF), the cause is unknown.
Physician-scientists at the University of Chicago are working to change that—because if you know the causes of disease, you are many steps closer to resolving it. This quest is particularly critical right now, because over the last three decades, mortality from ILD has doubled in the US.
UChicago, one of the few centers of excellence for treating the disease, draws many of the top people interested in studying it. A large team that included clinician-scientists, researchers, and radiologists from UChicago and NorthShore University HealthSystem, recently found some powerful indicators in predicting ILD outcomes—findings which will not only help physicians fine-tune treatments, but also provide clues of the disease’s mechanism.
One indicator involved the lymph nodes, the organs that 2018 Nobel Prize winner Jim Allison describe as like Rick’s Place in Casablanca: where all cells—the good guys, bad guys, reporters, and soldiers—go to hang out.
The team, led by Deji Adegunsoye, Jonathan Chung, Mary Strek, and Anne Sperling, found that when the lymph nodes tucked between the lungs were enlarged—a condition visible on a CT scan—patients had much worse lung function and significantly increased mortality.
They also found these patients had characteristic imbalances in cells and substances associated with the immune system, some of which could be found through a blood test. In fact, patients with large numbers of one specific interleukin—a protein generated by the immune system—circulating in their blood had the worst survival rates.
Taken together, these factors are likely biomarkers of the patient’s prognosis. Both can be monitored via minimally invasive testing, as opposed to the lung biopsies currently used.
Patients who show neither of these indicators likely have a truly different form of ILD, which not only means longer survival, but may also be differentially treatable with certain immunological drugs.
This latest research is only one strand of the many ways UChicago scientists are fighting ILD. The involvement of these immunological factors seems to corroborate what many team members have suspected through years of clinical work: that a variety of triggers—both chemical and microbial—may set up an immune response that attacks the lungs. IPF patients have often reported exposure to substances like organic solvents, stone dust, and/or mold. The Chicago team is looking for funding for a dedicated occupational health consultant to extensively document patients’ life histories in hopes of identifying disease triggers. As with a similar discovery that asbestos exposure causes the universally deadly mesothelioma, they hope to make ILP preventable.
The team is also working to significantly increase their biobank of lungs, which are exceptionally fragile and require a very specialized storage regimen. The lymph node findings were only possible because of the intense support of UChicago’s technical/biobanking staff (who must often respond on a moment’s notice when a patient undergoes a lung transplant) and strong collaboration with NorthShore. The team hopes UChicago can become a regional center of lung biobanking, to drive this kind of research.
And with the help of molecular engineers, the team is also working to develop microfluidic tools to drive that lung research and develop new drugs to address the immunological effects—tools and drugs that can be commercialized for use across the world, with the help of the Polsky Center for Entrepreneurship and Innovation.
For too long, interstitial pulmonary disease, especially IPF, have been a death sentence. It’s way past time for a change.
Elise Wachspress is a senior communications strategist for the University of Chicago Medicine & Biological Sciences Development office