
When I was a graduate student, my PI asked me how many μL of a particular antibody I used per million cells, and I told him my staining conditions were “per 100 μL” not “per million cells”. This triggered a heated debate about antibody amount – he argued that the amount of antibody needed was heavily dependent on cell number and I argued that it was more important to add antibody based on the staining volume than the cell number. I honestly don’t remember who “won” that argument that day, but given that he has superior debate skills and I lacked concrete evidence at the time I probably didn’t convince him of his error. Now that I’ve been working in a flow cytometry core facility for a few years, I’ve noticed that questions surrounding antibody concentration and cell number come up regularly, usually in the form of “how many cells should I stain?”. This question seems to be considered most often when researchers have a limited number of cells to study or they’re trying to scale up their staining protocol to sort a larger number of cells. Thinking back about my debate with my graduate school PI, I wondered how well researchers understand how cell staining conditions affect their results in order to properly interpret their results. In this post I’m going to discuss the impact of cell staining conditions on results and how to apply those concepts to determine how many cells are needed for an experiment.
Factors that Impact Cell Staining
Many factors during the cell staining process can change the resulting fluorophore intensity – including incubation time, incubation temperature, cell number, staining volume, and antibody concentration. In an ideal scenario, all factors should be kept exactly the same for all samples and controls. Realistically, this can be a challenge and sometimes these factors vary. So what happens when you vary the staining conditions? Of the five factors I mentioned, I’m going to assume that the incubation time and temperature will remain the same because these are the easiest parameters to keep constant and therefore should always be the same. Antibody concentration is completely dependent on staining volume, so I tend to lump those two together and always stain in 100 μL. Of course with large numbers of cells 100 μL may not be the best choice, but I’ll discuss that later. Knowing that it should be quite easy to keep incubation time, incubation temperature, and staining volume consistent between tubes and experiments, we are left with cell number and antibody concentration. These two factors can be the hardest to keep consistent, and I’d like to go into further detail about how changing these factors impacts the results.
Antibody Concentration
I will tell you now that antibody concentration is the most important factor in the cell staining protocol. If the antibody concentration changes, the difference in staining pattern can be drastic. Because of this the I strongly recommend titrating all antibodies and making a master mix of all antibodies to stain all samples. Titration will determine the optimal amount of antibody to use and a master mix will ensure that all samples get the same amounts of antibodies and reduce pipetting error. For reliable results I don’t recommend pipetting less than 2 μL. If small volumes are required, make a dilution of the antibody first! Figure 1 demonstrates how antibody concentration can affect the resolution of the positive population. In the figure, the best resolution is determined by the highest staining index.
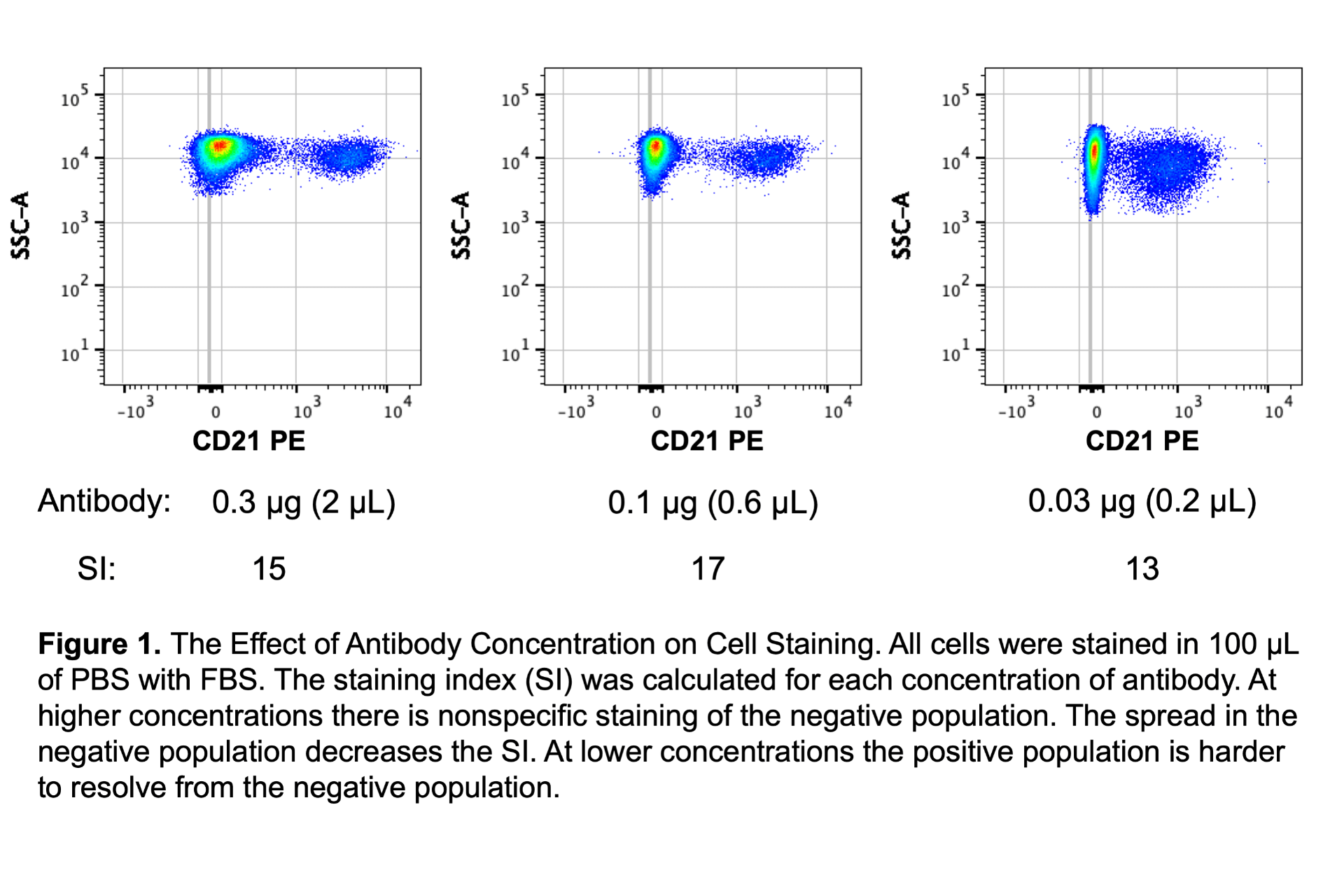 Since antibody concentration is so critical, another tip is to keep track of the amount of antibody in mg/mL as opposed to the volume of antibody per volume of staining buffer. The reason for this has to do with simplifying titrations if the fluorophore is changed for a particular antibody clone. If I know that I’ve titrated a PE antibody to 0.1 μg in 100 μL and I decide to switch the fluorophore to FITC (same antibody clone), then I know that I easily calculate and use 0.1 μg in 100 μL even though the PE antibody has a concentration of 0.2 mg/mL and the FITC antibody has a concentration of 0.5 mg/mL. If I keep track of my antibody titrations as “1:100” or “2 μL”, then it is more work to switch fluorophores on antibodies. To be clear, the best practice is always to titrate every single antibody, but using the same mg/mL (or μg per 100 μL) often works and the approach of titrating each antibody clone regardless of fluorophore is better than not titrating at all.
Since antibody concentration is so critical, another tip is to keep track of the amount of antibody in mg/mL as opposed to the volume of antibody per volume of staining buffer. The reason for this has to do with simplifying titrations if the fluorophore is changed for a particular antibody clone. If I know that I’ve titrated a PE antibody to 0.1 μg in 100 μL and I decide to switch the fluorophore to FITC (same antibody clone), then I know that I easily calculate and use 0.1 μg in 100 μL even though the PE antibody has a concentration of 0.2 mg/mL and the FITC antibody has a concentration of 0.5 mg/mL. If I keep track of my antibody titrations as “1:100” or “2 μL”, then it is more work to switch fluorophores on antibodies. To be clear, the best practice is always to titrate every single antibody, but using the same mg/mL (or μg per 100 μL) often works and the approach of titrating each antibody clone regardless of fluorophore is better than not titrating at all.
Cell number
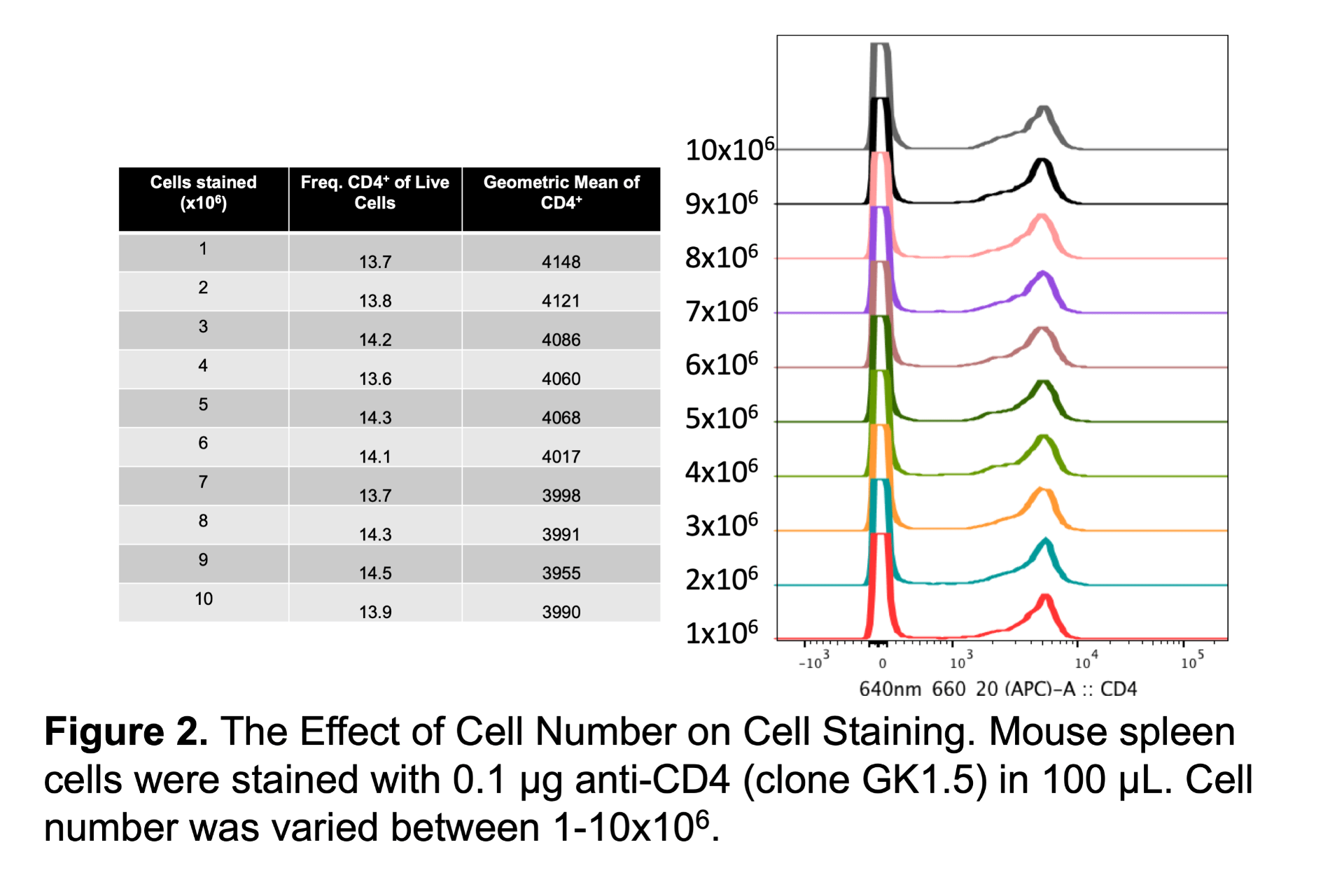
Some people may be surprised to find out that cell number can have little effect on the data. In the example below I stained between 1-10 million cells with the same concentration of anti-CD4 antibody (Figure 2). Except for cell number all other staining conditions were kept the same. Even with a tenfold increase of cells there is no significant impact on the frequency or MFI of the positive population. Note that this may not be true for every single antibody because some antibodies are more sensitive to cell number than others, but in general cell number is not as critical as other staining conditions. In practice this means that a single antibody concentration can work for a range of cell concentrations.
How to apply this to your flow cytometry experiments
Now that we have good understanding of how cell number and antibody concentration impact results, we can address the question of how many cells should be stained for an experiment. Except that we still need some more information because the answer depends on the specifics of each individual experiment. When researchers ask me how many cells are needed for their experiment I typically need to know two things: how many cells can the researcher obtain from each sample and how rare is the smallest population of interest. When cell number is limited, researchers need to think more carefully about how many cells to use for controls and how changing the cell number impacts the staining of each marker in the panel. To address the question of “how many cells should be stained” I’ll break it down into three separate different questions:
- How many cells should be stained for the actual sample?
- How many cells should be stained for the controls?
- How many cells should be stained when titrating an antibody?
The first question is quite simple. By default, we suggest staining 1×106 cells. However, the number of total cells is best determined by the frequency of the rarest population of interest. Let’s say we want to examine a blood sample that contains 50% neutrophils, 10% monocytes, 30% lymphocytes, and 0.5% regulatory T cells (Tregs). If the focus of the experiment is on the monocytes as the rarest population of interest, we don’t need a large number of cells. Knowing that the monocytes are about 10% of the total cells, we can calculate that collecting 50,000 events will give us about 5,000 monocytes. If the sample is not limited, I recommend staining 0.5×106-1×106 cells. However, if the sample is limited, I can calculate that I would be able to stain 1×105 cells and still be able to analyze an appropriate number of cells. Keep in mind that cells are lost in the staining process and pushing sample through the cytometer, so don’t stain 1×105 cells and expect to be able to analyze exactly 1×105 cells!
Now let’s change the focus of the experiment – let’s say the goal is to examine the Tregs. Based on the knowledge that the Tregs are only 0.5% of the total cells, only 250 Tregs could be analyzed in a total of 50,000 cells. It is recommended to have at least 100-500 events in the rarest population, depending on how distinct the markers are on the population. In this scenario, it is possible that 50,000 total cells could be used to analyze Tregs, but more cells would certainly be a good idea.
The second and third questions regarding controls and titrations are a bit more difficult to answer and boils down to how many cells are available. If the sample amount is not limited, then the same number of cells should be used to stain the controls and the sample. If the sample amount is limited, then may be possible to stain fewer cells for the controls to maximize the number of cells in the fully stained sample. The abundance of populations in the sample should also be taken into consideration for fluorescence minus one (FMO) controls. If the FMO is needed for a rare population of cells, then the same number of cells should be stained and analyzed for the sample and FMO control. If the marker is higher up on the gating tree and therefore used for a larger percentage of cells, it may be possible to get by staining fewer cells – assuming that the same staining intensity is achieved with a lower concentration of cells. If it is difficult to obtain enough cells for both the sample and the controls, one solution is to determine if compensation beads can be used instead of single stained cells. However, compensation beads are not always as good as cells and cells will always be needed for FMO controls.
If it is determined that fewer cells can be used for controls compared to the sample, the staining conditions should be considered. Often times one concentration of antibody could be used for a range of cell concentrations, so the same antibody amount and staining volume may be acceptable. However, if there is a large difference in cell number in the sample and the control or if an antibody is particularly sensitive to cell number, then better results can be achieved if the antibody concentration and cell concentration are the same. For example, if the control is stained with 0.25×106cells and 1 μL of antibody in 100 μL we can keep the antibody and cell concentrations the same by staining 5×106cells and 20 μL of antibody in 2 mL. However 20 μL is a lot of antibody, so it certainly cheaper to consider staining 5×106cells in 10 μL of antibody in 1 mL or even 5 μL of antibody in 0.5 mL. The important thing is to keep the antibody concentration exactly the same and then keep the cell concentration similar between the controls and samples, knowing that a range of cell concentrations can work for a single antibody concentration. When scaling up a staining protocol, the best practice would be to stain two tubes—the normal and scaled up number of cells—in the same experiment and determine if the staining patterns are comparable.
Another common scenario where cell number becomes a critical discussion point is transitioning from a benchtop analyzer to a sorting experiment. For example, after analyzing 1×106 cells on a benchtop analyzer and finding a really interesting population, the next step may be to sort that population. But in order to get enough cells for the experiment, let’s say that 10×106 cells are needed based on the known frequency of the interesting population and the number of cells needed for the post-sort experiment. The staining protocol be scaled up for the sorting experiment similar to what I described above. It is unlikely that ten times as much antibody needed to stain ten times as many cells! I typically make an assumption that titrated antibodies work for up to 5×106 cells, so as an example I would recommend scaling up the original conditions of 0.1 mg of antibody and 1×106 cells in 100 mL to 0.2 mg of antibody and 10×106 cells in 200 mL. However, this is a generalization and scaling up a staining protocol in this way probably won’t work for every antibody and every cell type. The best way to figure it out is to test it and compare to the original protocol!
The key to any good science experiment is consistency – use the same reagents, protocol, and instrument and real results will be reproducible. But there are times when scientists aren’t able to be as consistent as they want to be. As I discussed, there may not be enough cells for all of the controls or cost of reagents may become a limiting factor. In these scenarios, a good flow cytometrist will understand their staining protocol and how the cell concentration and staining conditions may be altered without compromising the results. And a good graduate student will prove their PI wrong with solid data. (I may be a few years late and he might not read this, but I think an “I told you so” is in order!)
Looking for more flow cytometry resources?

Avoid Bad Data Series
This popular series of blog posts discusses how to identify bad flow cytometry data and provides tips for avoiding common mistakes.

Flow Basics 2.0
A comprehensive online course that covers the protocol and optimization of staining cells, panel design, choosing controls, and instrument setup.

Sample Preparation
Learn about flow cytometry staining protocols, antibody titration, fixation considerations, etc.
Hello Laura,
My name is Cesar De Jeronimo, a physiology doctorate student at Penn State University. I’ve come across your article and found the information quite useful in my flow cytometry experiments. I wanted to reach out for some advice. Is there any tips and guidance you can provide for attempting to process 100K cells for intracellular and extracellular staining? Currently I am attempting to characterize infiltrating macrophages within myocardium after ischemic injury and the total cells I regularly isolate are around 1×10^6, thus have limited cells to run my controls and experimental samples. The intracellular staining process is what causes me to lose my samples as when I’ve done extracellular staining only, the sample is retained. Thank you for your time.
Hi Cesar,
You could use compensation beads or another cell type for you single stain controls (though there are pros and cons to doing that). Alternatively, there might be something in your protocol that’s causing you to lose cells. If you would like, you can email me at LJohnston@bsd.uchicago.edu to go over our consultation services. I would need more information about your experiment to fully address your question, so a consultation would be best.
Very useful. Thank you!
Hello Laura
My name is Fabiola. An Immunology Ph.D. student. Very interesting your article. I have some questions about titration when my cells have low expression of molecules like T-bet, FOXP-3. How can I do my titration of these antibodies?. I appreciate your help. Thank you.
Hi Fabiola,
I’m not sure if you’re using “low expression” to refer to the amount of marker per each individual cell or the percentage of positive cells within the entire sample. All basic guidelines for titrating apply to all types of markers (low and high expression/frequency). Ideally, you should titrate all antibodies using the full staining protocol you plan to use for your experiment and use the same tissue as your experiment. If you are using a treatment/model/disease, it is best if you can use those exact cells for the titration, especially if you expect there to be higher expression of a marker in a treated condition vs a control condition. If your marker is only expressed on a small fraction of cells (maybe 0.5% of the total cells), you may find it useful to include other markers to allow you to gate on specific cell subsets. Adding identifying markers to your titration tube can also be useful for low expressed markers (molecules per each cell) to help identify that staining is not nonspecific, though that is not the only way to identify nonspecific staining. For example if you have transcription factors that you expect to be restricted to T cells and not expressed by B cells, you may want to include a T cell marker (like CD3) and B cell marker (like CD19) while titrating your transcription factors. Then you can analyze the titration data by gating on either the CD3+ cells or CD19+ cells and ensure you are choosing a concentration of antibody where there is no background staining on the B cells.
A less ideal option would be to use a different cell line or tissue type that you are certain has good expression of your marker and do the titration on these cells. If you use this approach I would still recommend testing out the chosen concentration on your actual tissue of interest before you do an entire experiment.
Hi Laura!
Very useful points!
I had a question regarding the balance between cells number and the volume of the master mix. We are staining for CD29+, CD34+ progenitor cells in the adipose tissue. And, they are said to consist almost 70% of the purified cells. We have previously titrated the antibodies. Our technician has stained 5 million cells all in one well with 50ul master mix and she was not able to detect high enough signals, and no double positive population was captured. Do you think this has happened because the balance between cell number and master mix volume was not correct?
Hi Mahshid,
It’s a bit challenging for me to troubleshoot this without having a conversation with you, but from what understand it sounds like you titrated antibodies successfully using one set of staining conditions and then changed those staining conditions to stain 5 million cells with 50uL of master mix. I would suggest staining your cells using the exact staining conditions used for the successful titration experiments. If you can be successful with that, then you could choose to change one thing. For example, maybe you originally stained 1 million cells. Go back to staining 1 million cells and make sure that’s working as expected. If it is then you might choose to change one thing- maybe you switch to 5 million cells. Then you can confirm if that change makes a difference or not. But you should make sure you only change one of the staining conditions (time, temp, antibody concentration, cell concentration, staining volume) at a time, otherwise you’ll have trouble determining what caused the issue.
Hi Laura,
my name is Giulia, I am a postdoc researcher in immunology.
First of all, thank you very much for what you took the time to write, very useful!
I would like to submit you my staining concern and how I think to proceed to have your useful opinion on this.
So, let’s imagine I titrate my Abs with 1×10^6 cells in 100ul of staining buffer and I find that the best concentration to use my Ab is 1ug(1ul) Ab (so a final dilution of 1:100 if the initial concentration of my Ab is 1mg/ml).
Now, I need to proceed to the sorting of some very rare populations (the stromal cells) from the murine bone marrow. These cells represent 0.1-0.2% of the total cells and so if I normally get 100×10^6 cells in total from the bone marrow I will have around 100.000-200.000 of these cells (that I need all for the sorting). To have the correct amount of Abs is crucial for me in this experiment as my sorting will be based on negative selection and so I really want all positive cells for a certain antigen to be stained for it with the Ab. So I will have to stain 100×10^6 cells with the Ab that I have titrated above. If we assume that 1ul of Ab was good for 1×10^6 cells we can make the assumption (as you said that titrated antibodies work for up to 5×10^6 cells) that it can be good up to 5×10^6 then I will stain 10×10^6 cells with 2ul in 200ul (this is also the example you gave, to maintain the Ab concentration the same which is the most important parameter). Now, can I consider that 2ul in 200ul will be fine to stain up to 50×10^6 cells (always based on the assumption that you mentioned )? And so stain 100×10^6 cells with 3ul of Ab in 300ul of staining buffer? Although, for such a big amount of cells (100×10^6 cells) I would stain in maybe 500ul (and so with 5ul of the Ab) for the cells not to be too concentrated during the staining. What do you think about this?
However, my very strong doubt is that in the example I gave you I am assuming that the ratio quantity of Ab/number of cells follow and exponential curve (which I hardly doubt). Indeed, it is maybe more correct to think that 1ul of Ab in 100ul of staining buffer would stain well from 1-10×10^6 cells, so adding 1ul (in 100ul of staining beffer..so having at the end 2ul in 200ul) of Ab is ok to stain 10-20^6 cells, and so on so adding 1ul more (and so having in total 3ul of Ab in 300ul of staining buffer) is ok to stain 20-30^106 cells (so following a linear proportion instead of an exponential one…so lets say at the end saying that 1ul of Ab is good to stain 1-10^6 of cells and so that each 10^6 cells more we have we have to add 1ul more). So at the end to stain 100^10^6 cells with 10ul of Ab in 1ml of final staining…is this correct? Honestly…I never saw someone staining in such big final volume..would it be the same to do 10ul of Ab in 500ul of final staining buffer?
Also, what you show in Figure 2 is different as compared to when you say “I typically make an assumption that titrated antibodies work for up to 5×10^6 cells” because, if the Ab was titrated with 1×10^6 cells with 1ul Ab in 100ul of staining according to Figure 1 this should work up to 1×10^6 cells, right? Can you help me understanding which would be the more correct assumption?
I thank you a lot in advance for your help really,
You cannot even imagine how much you can help with your suggestions,
You are offering a very important service for the scientific community so thank you thank you!
Best
Hi Giulia,
Your question started a debate in the core. I’d say that you’ll absolutely get separation between the positive and negative cells if you ramp up the antibody amount linearly with the staining volume – so in this case, starting at 1ul Ab in 100ul of staining volume, you’d go with 50ul Ab in 5ml staining volume for your 100 million cells. But from I’ve seen, you’ll likely be way beyond saturation point and might just be wasting antibodies. Here’s what I suggest: when you get your 100 million cells for sorting, split the sample and do a 3 point titration. Resuspend at 20million cells/ml, your highest concentration will be 1:100 and find two lower points that you like. You’ll be set for your following experiments.