By Rob Mitchum // October 31, 2014
While genomic sequencing gets cheaper, more extensive, and more routine, the analysis of those sequences remains a difficult challenge. Clinics and laboratories studying genetics use computational methods and bioinformatics pipelines to streamline this process, converting raw sequencing results into accurate and actionable insights. In a new collaboration between Computation Institute and University of Chicago Medicine researchers, one key step of genome analysis was improved by instituting a form of computational democracy, combining the power of four different algorithms to produce more reliable results.
Geneticists seeking the footprints of disease in DNA increasingly look for rare variants — mutations, deletions, or insertions associated with a disease that appear in a small number of individuals or families. But finding these variants is the proverbial needle in a haystack problem, requiring deeper sequencing of patients (to the exome or even whole-genome level) and “variant calling” algorithms that exhaustively comb through the sequences for potentially relevant gene variations. If a variant caller is too selective, it could miss an important variant; too loose, and it could give researchers far more candidates than they would be able to further test for biological or clinical significance.
“Some callers take a “brute force” approach and are very sensitive while others try to integrate multiple quality measures at each position or information from nearby variants to decide if a new variant is real or not,” said Lea Davis, research associate in the Department of Medicine and first author of the paper. “When you only use one algorithm, it’s hard to know what you’re not catching. Alternatively, if you use something that is very sensitive, but not very specific, you may get a lot of false positives.”
Given this tricky task, several different groups have written their own variant calling algorithms, each using slightly different strategies for sniffing out targets, each with their own strengths and weaknesses. To find the best balance, scientists from the laboratory of CI Senior Fellow Nancy Cox worked with the Globus Genomics team to borrow a strategy from other computational fields: ensemble modeling.
Commonly used for weather forecasting and machine learning, ensemble methods involve using multiple algorithms or multiple starting conditions on data and combining the results to achieve higher accuracy or observe a range of possible outcomes. For genomics, the Globus/UChicago team constructed the Consensus Genotyper for Exome Sequencing (CGES), an ensemble method that combines the perspectives of four variant-calling algorithms to get higher quality results. A paper describing and demonstrating the method was published in September by Bioinformatics.
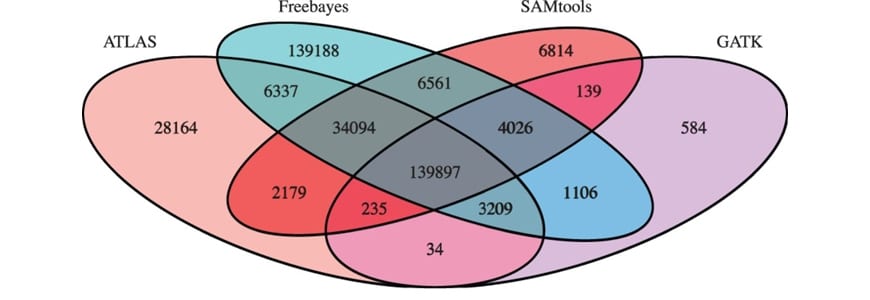
The system works like a board taking a vote. Each of the four variant callers goes over the sequence independently and identifies variants using its particular criteria. The pipeline then combines the results from the four algorithms and produces three different outputs for further analysis. The consensus calls are the unanimous variants, detected by all four callers. Partial consensus calls are those that achieved a majority, labeled as a variant by at least three algorithms. Users can also take away the union of all calls — every single variant identified by at least one algorithm.
“Each caller uses its own bias in calling the variant, so when you use multiple algorithms, you’re in a way using them to control for bias,” said Ravi Madduri, co-author and software engineer for Globus. “When all four call a particular variant, it has a high probability that the significance of the variant is high.”
The different outputs present a gradient of options for further analysis, depending on a scientist’s needs. The consensus set will include the fewest variants, but with the highest accuracy. The set of every variant identified by at least one algorithm will have the most variants, which may be useful if researchers are searching for very rare or hard-to-find sequences.
To verify these qualities, the researchers used CGES on genetic sequences from 132 autistic individuals and their family members, provided by Vanderbilt University and the University of Illinois at Chicago. As expected, the consensus set contained the fewest variants; though still an imposing number at nearly 140,000. When compared to Sanger sequencing — the gold standard method for high-resolution genome sequencing — the CGES method was 96 percent accurate, an improvement of 2 to 7 percent over the individual callers alone.
“A two percent difference may not seem like much, but it can mean the difference between hundreds of misclassified variants,” Davis said. “One example of how this improved performance will translate to better results is in the search for de novo variation, or variation that appears in a child but not in their parents. These variants can be very difficult to find because on the surface there is no way to tell a de novo variant from a false positive call in the child or a false negative call in the parents. By using an ensemble approach, you cut down on the number of false positives in kids, and by checking against the union of all calls in parents you cut down on the number of false negatives.”
After proving its success, the creators of CGES wanted to make sure that it would be easily accessible to the broader genetics community. All code for the method was posted to Github, so that other users can use and improve the algorithm. CGES will also be introduced into Galaxy, an online suite of bioinformatics tools, and incorporated into Globus Genomics, which combines Galaxy with cloud-based data transfer and computing resources. Already, the Cox lab has applied the method to drug response phenotypes, and the laboratory of CI Senior Fellow Funmi Olopade has used CGES for studying breast cancer cases from Nigeria.
“We didn’t develop CGES just for one experiment,” said Madduri, “A lot of big analysis tools are developed but just disappear after the paper was written. The pipeline that we created is useful, and we know that it can be used in other areas.”
Besides Davis, Madduri, and Cox, other authors on the paper include Alex Rodriguez, Uptal Davé, and Ian Foster of the Computation Institute, Vassily Trubetskoy of the University of Chicago, Emily Crawford and James Sutcliffe of Vanderbilt University, and Edwin Cook of the University of Illinois at Chicago.